Task aliasing
When you need to call a task more than once in a workflow, use task aliasing.
It would be tedious to copy-paste a task's definition and change the name each time you needed to use it again in the workflow. This method, termed copy and paste programming, is simple enough up front but difficult to maintain in the long run. Imagine you found a typo in one of your tasks--you'd need to fix that typo in every single pasted task! However, using WDL's built-in task aliasing feature, you can call the same task code and assign it an alias. Then, following the principle of hierarchical naming, in order to access the output of an aliased task we use the alias, rather than the original task name.
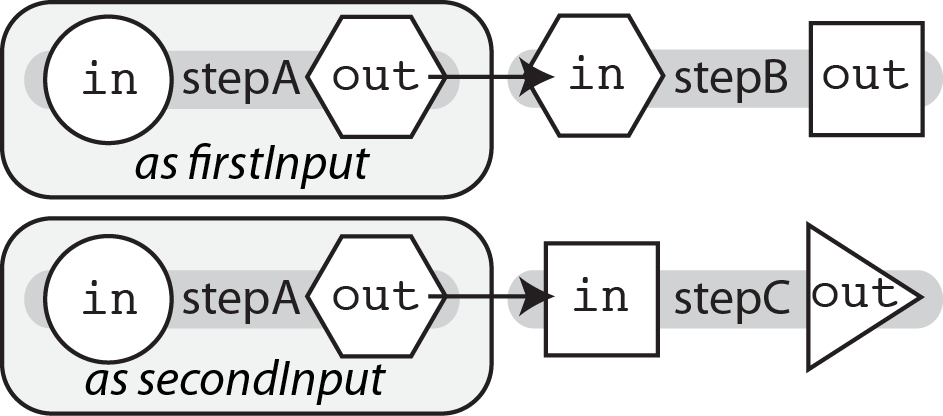
To use an alias, we use the syntax call taskName as aliasName, as demonstrated in the code snippet below:
call stepA as firstSample {
input:
in = firstInput
}
call stepA as secondSample {
input:
in = secondInput
}
call stepB {
input:
in = firstSample.out
}
call stepC {
input:
in = secondSample.out
}
Generic example script
The workflow and its tasks altogether in a WDL script would look as follows:
version 1.0
workflow taskAlias {
input {
File firstInput
File secondInput
}
call stepA as firstSample {
input:
in = firstInput
}
call stepA as secondSample {
input:
in = secondInput
}
call stepB {
input:
in = firstSample.out
}
call stepC {
input:
in = secondSample.out
}
}
task stepA {
input {
File in
}
command <<<
programA I=~{in} O=outputA.ext
>>>
output {
File out = "outputA.ext"
}
}
task stepB {
input {
File in
}
command <<<
programB I=~{in} O=outputB.ext
>>>
output {
File out = "outputB.ext"
}
}
task stepC {
input {
File in
}
command <<<
programC I=~{in} O=outputC.ext
>>>
output {
File out = "outputC.ext"
}
}
Concrete example
Let's take a look at this task aliasing concept inside a real-world example; using the GATK, this workflow separates SNPs and Indels into distinct vcf files using the same task, select, but two distinct aliased calls, selectSNPs and selectIndels. The distinct outputs of these calls are then hard filtered by separate tasks designed specifically for them, hardFilterSNP and hardFilterIndel, respectively.
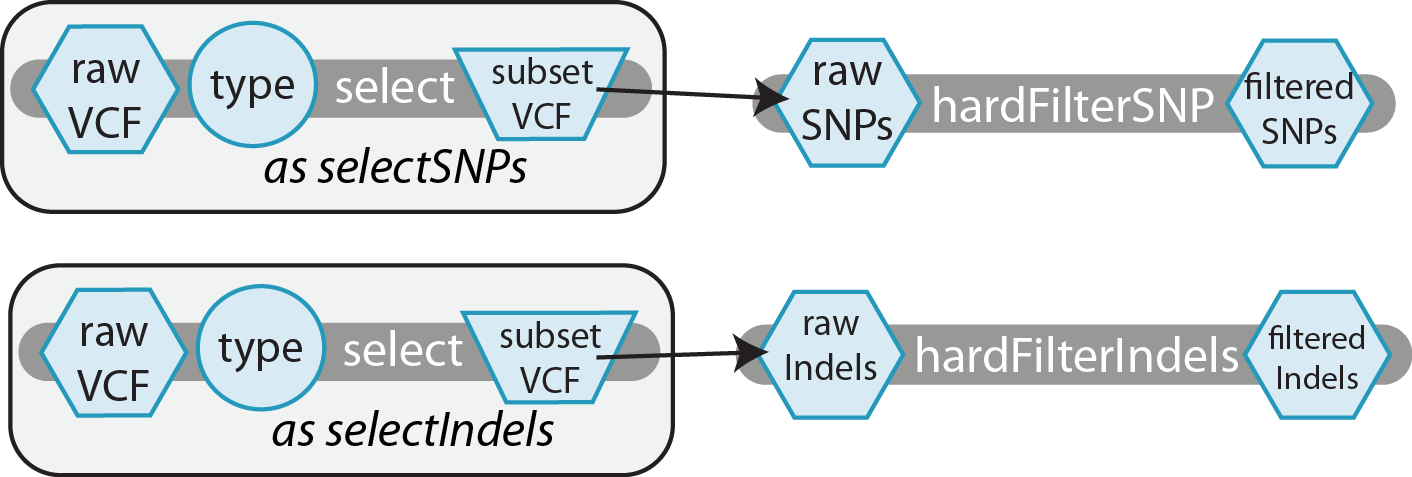 For this toy example, we have defined three tasks:
For this toy example, we have defined three tasks:
- select takes in a
String type, specifying "SNP" or "Indel", and aFile rawVCF, outputting aFile rawSubsetthat contains only variants of the specified type. - hardFilterSNP takes in a
File rawSNPsand outputs aFile filteredSNPs. - hardFilterIndelstakes in a
File rawIndelsand outputs aFile filteredIndels.
Concrete example script
The above workflow description would look like this in its entirety:
version 1.0
workflow taskAliasExample {
input {
File rawVCFSample
}
call select as selectSNPs {
input:
type = "SNP",
rawVCF = "rawVCFSample"
}
call select as selectIndels {
input:
type = "INDEL",
rawVCF = "rawVCFSample"
}
call hardFilterSNP {
input:
rawSNPs = selectSNPs.rawSubset
}
call hardFilterIndel {
input:
rawIndels = selectIndels.rawSubset
}
}
task select {
input {
String type
File rawVCF
}
command <<<
java -jar GenomeAnalysisTK.jar \
-T SelectVariants \
-R reference.fasta \
-V ~{rawVCF} \
-selectType ~{type} \
-o raw.~{type}.vcf
>>>
output {
File rawSubset = "raw.~{type}.vcf"
}
}
task hardFilterSNP {
input {
File rawSNPs
}
command <<<
java -jar GenomeAnalysisTK.jar \
-T VariantFiltration \
-R reference.fasta \
-V ~{rawSNPs} \
--filterExpression "FS > 60.0" \
--filterName "snp_filter" \
-o .filtered.snps.vcf
>>>
output {
File filteredSNPs = ".filtered.snps.vcf"
}
}
task hardFilterIndel {
input {
File rawIndels
}
command <<<
java -jar GenomeAnalysisTK.jar \
-T VariantFiltration \
-R reference.fasta \
-V ~{rawIndels} \
--filterExpression "FS > 200.0" \
--filterName "indel_filter" \
-o filtered.indels.vcf
>>>
output {
File filteredIndels = "filtered.indels.vcf"
}
}
For simplicity, we omitted the handling of index files, which has to be done explicitly in WDL.