Scatter-gather parallelism
Parallelism is a way to make a program finish faster by performing several operations in parallel, rather than sequentially (i.e. waiting for each operation to finish before starting the next one). For a more detailed introduction on parallelism, you can read about it in-depth here.
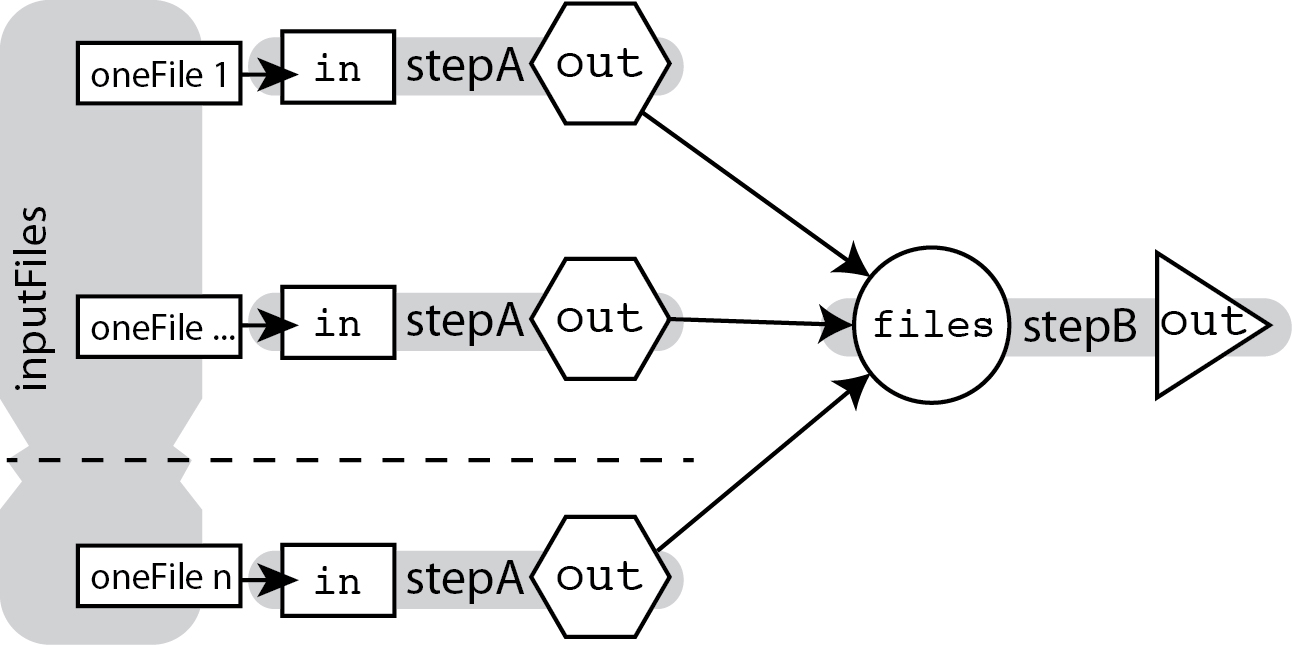
To do this, we use the scatter function described in the WDL 1.0 spec, which will produce parallelizable jobs running the same task on each input in an array, and output the results as an array as well.
The code below shows an example of the scatter function:
version 1.0
workflow wf {
input {
Array[File] inputFiles
}
scatter (oneFile in inputFiles) {
call stepA {
input:
in = oneFile
}
}
call stepB {
input:
files = stepA.out
}
}
The magic here is that the array of outputs is produced and passed along to the next task without you ever making an explicit declaration about it being an array.
Even though the output of stepA looks like a single file based on its declaration, just referring to stepA.out in any other call statement is sufficient for WDL to know that you mean the array grouping the outputs of all the parallelized stepA jobs.
In other words, the scatter part of the process is explicit while the gather part is implicit.
Generic example script
To put this in context, here is what the code for the workflow illustrated above would look like in full:
version 1.0
workflow ScatterGather {
input {
Array[File] inputFiles
}
scatter (oneFile in inputFiles) {
call stepA {
input:
in = oneFile
}
}
call stepB {
input:
files = stepA.out
}
}
task stepA {
input {
File in
}
command <<<
programA I=~{in} O=outputA.ext
>>>
output {
File out = "outputA.ext"
}
}
task stepB {
input {
Array[File] files
}
command <<<
programB I=~{files} O=outputB.ext
>>>
output {
File out = "outputB.ext"
}
}
Concrete example
Let’s look at a concrete example of scatter-gather parallelism in a workflow designed to call variants individually per-sample (HaplotypeCaller), then to perform joint genotyping on all the per-sample GVCFs together (GenotypeGVCFs).
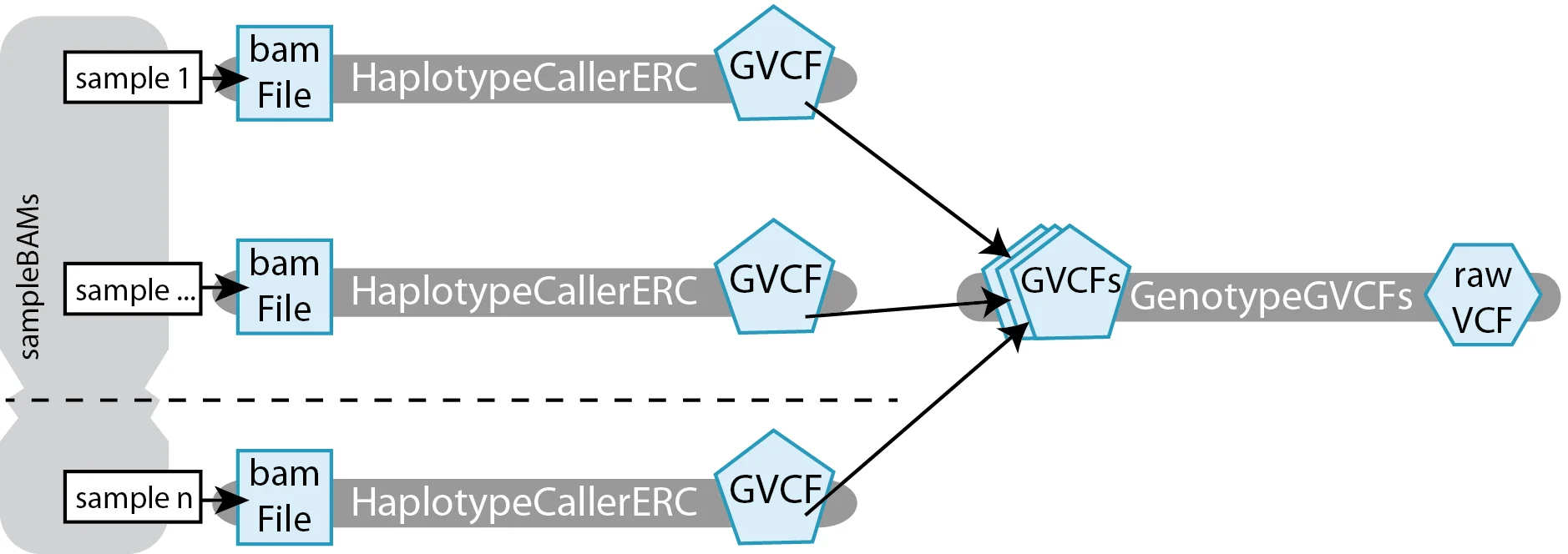
The workflow involves two tasks:
HaplotypeCallerERCtakes in aFile bamFileand produces aFile GVCF.GenotypeGVCFstakes in anArray[File] GVCFand produces aFile rawVCF.
Concrete example script
This is what the code for the workflow illustrated above would look like:
version 1.0
workflow ScatterGatherExample {
input {
Array[File] sampleBAMs
}
scatter (sample in sampleBAMs) {
call HaplotypeCallerERC {
input:
bamFile=sample
}
}
call GenotypeGVCF {
input:
GVCFs = HaplotypeCallerERC.GVCF
}
}
task HaplotypeCallerERC {
input {
File bamFile
}
command <<<
java -jar GenomeAnalysisTK.jar \
-T HaplotypeCaller \
-ERC GVCF \
-R reference.fasta \
-I ~{bamFile} \
-o rawLikelihoods.gvcf
>>>
output {
File GVCF = "rawLikelihoods.gvcf"
}
}
task GenotypeGVCF {
input {
Array[File] GVCFs
}
command <<<
java -jar GenomeAnalysisTK.jar \
-T GenotypeGVCFs \
-R reference.fasta \
-V ~{GVCFs} \
-o rawVariants.vcf
>>>
output {
File rawVCF = "rawVariants.vcf"
}
}
For simplicity, we omitted the handling of index files in the code above, which has to be done explicitly in WDL.
Additionally, please note that we did not expressly handle delimiters for Array data types in this example.
Scatter limitations
Note that the max allowable scatter width per scatter in Terra is 35,000.