Branch and merge
The ability to connect inputs and outputs described in the Linear chaining and Multiple I/O chapters can be further extended to direct a task's outputs to separate computation paths and merge the results together. This technique, known as branching and merging, is a common way to run multiple steps of a broader analysis in parallel.
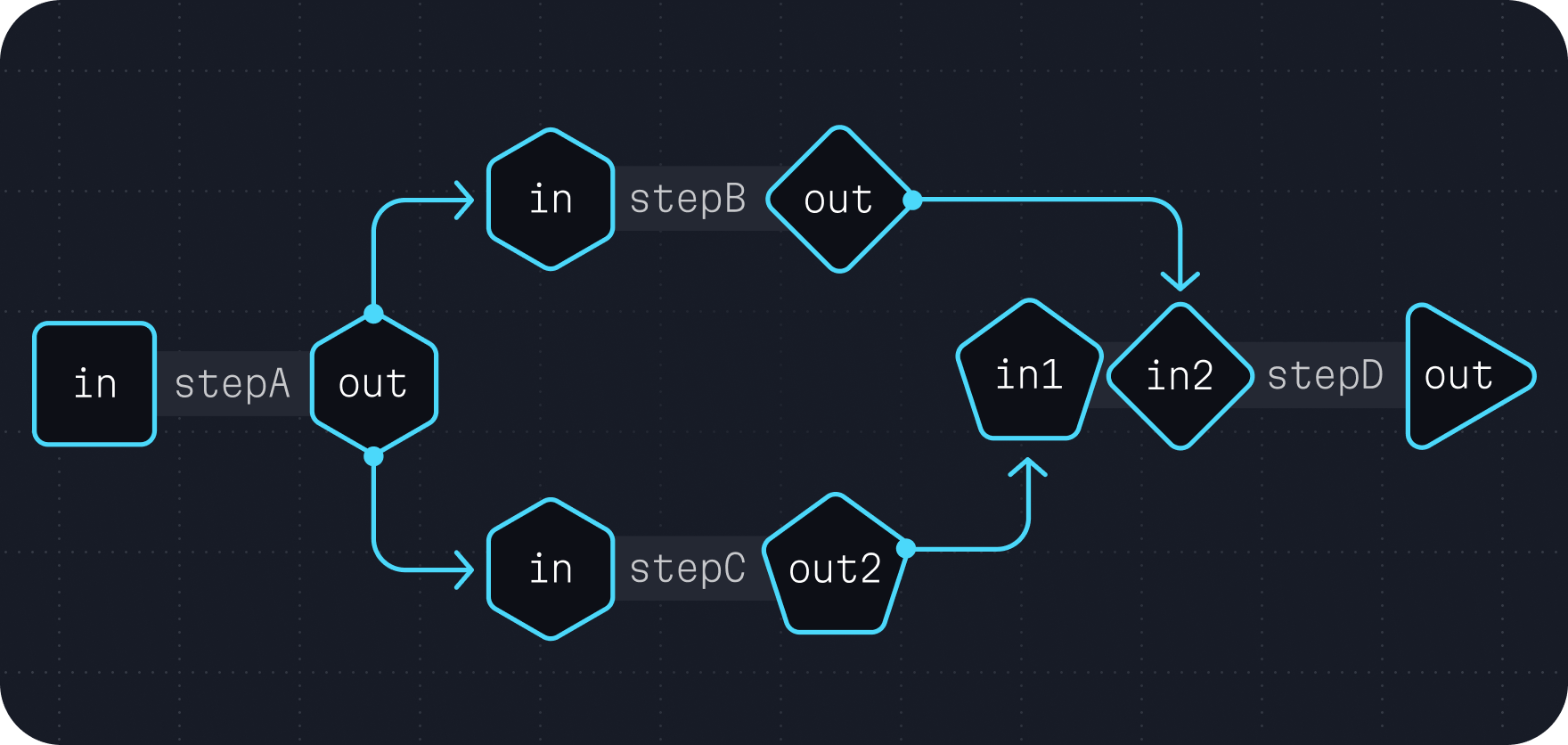
This often takes a form similar to the following example.
wdl
# ... task definitions ...
workflow run {
# Run the first task.
call stepA {}
# Run `stepB`, connecting the `in` input to `stepA`'s `out` output.
call stepB { input: in = stepA.out }
# Run `stepC`, connecting the `in` input to `stepA`'s `out` output.
call stepC { input: in = stepA.out }
# Run `stepD`, connecting the `in1` input to `stepB`'s `out` output
# and the `in2` input to `stepC`'s `out` output.
call stepD { input: in1 = stepB.out, in2 = stepC.out }
}Continuing our example
To build on the example started in Multiple I/O, we'll extend this workflow to have a filtering step for single nucleotide polymorphisms and indels separately. This introduces a branch in our computation graph where the filtering steps can be run concurrently and merged back together after completion.
wdl
version 1.3
# (1) We write a task to split the VCF into multiple VCFs.
task split_vcf {
input {
File vcf
}
command <<<
picard SplitVcfs \
I="~{vcf}" \
SNP_OUTPUT="snp.vcf" \
INDEL_OUTPUT="indel.vcf" \
STRICT=false
>>>
output {
File snp_vcf = "snp.vcf"
File indel_vcf = "indel.vcf"
}
}
# (2) We write a task to filter SNPs using our criteria.
task filter_snps {
input {
File vcf
}
command <<<
gatk VariantFiltration \
-V ~{vcf} \
-O filtered.vcf \
--filter-expression "QD < 2.0 || FS > 60.0 || MQ < 40.0 || MQRankSum < -12.5 || ReadPosRankSum < -8.0" \
--filter-name "snp_filter"
>>>
output {
File filtered_vcf = "filtered.vcf"
}
}
# (3) We write a task to filter INDELs using out criteria.
task filter_indels {
input {
File vcf
}
command <<<
gatk VariantFiltration \
-V ~{vcf} \
-O filtered.vcf \
--filter-expression "QD < 2.0 || FS > 200.0 || ReadPosRankSum <-20.0" \
--filter-name "indel_filter"
>>>
output {
File filtered_vcf = "filtered.vcf"
}
}
# (4) We write a task to merge these two VCFs.
task merge_vcfs {
input {
File snp_vcf
File indel_vcf
}
command <<<
picard MergeVcfs \
I="~{snp_vcf}" \
I="~{indel_vcf}" \
O="combined.vcf"
>>>
output {
File combined = "combined.vcf"
}
}
# (5) We write a workflow that branches to the two filtering steps
# and merges back in the `merge_vcfs` task.
workflow run {
input {
File vcf
}
call split_vcf { input: vcf }
# (a) The branch happens here.
call filter_snps { input: vcf = split_vcf.snp_vcf }
call filter_indels { input: vcf = split_vcf.indel_vcf }
# (b) The merge happens here.
call merge_vcfs { input:
snp_vcf = filter_snps.filtered_vcf,
indel_vcf = filter_indels.filtered_vcf,
}
output {
File combined = merge_vcfs.combined
}
}