Conditionals (if/else)
Conditionals have been implemented as of Cromwell version 24
Sometimes when pipelining, there are steps you want to run sometimes and not other times. This could mean switching between two paths (e.g. run a tool in modeA vs. run a tool in modeB) or skipping a step entirely (e.g. run a tool vs. not running a tool). In cases such as these, we will use a conditional statement.

To use a conditional statement in WDL, you write a standard if()statement:
if (shouldICallStepB) {
call stepB {
input:
in = stepA.out
}
}
The if() statement can be controlled explicitly, as we have done in the above example by using a Boolean variable. It can also be controlled implicitly by testing the value of some other variable that has its own purpose besides being a switch mechanism. (i.e. if (myVar>0) { call stepB {} }).
Handling the output of a conditional step is a bit different; see the Generic example script below for details on StepC.
One thing WDL does not yet have is an else() function. Right now to get around that, we write paired if() statements using the !modifier to get the opposite value of the original variable, like in the code example below:
input {
Boolean myBoolVar
}
if (myBoolVar) {
call taskA {}
}
if(!myBoolVar) {
call taskB {}
}
Generic example script
Take a look at this example WDL in the code below:
version 1.0
workflow Conditional {
input {
File firstInput
Boolean shouldICallStepB
}
call stepA {
input:
in = firstInput
}
if (shouldICallStepB) {
call stepB {
input:
in = stepA.out
}
}
call stepC {
input:
in_maybe = stepB.out
}
}
task stepA {
input {
File in
}
command <<<
programA I=~{in} O=outputA.ext
>>>
output {
File out = "outputA.ext"
}
}
task stepB {
input {
File in
}
command <<<
programB I=~{in} O=outputB.ext
>>>
output {
File out = "outputB.ext"
}
}
task stepC {
input {
File? in_maybe
}
command <<<
programB I=~{in_maybe} O=outputB.ext
>>>
output {
File out = "outputB.ext"
}
}
It is important to note that stepC’s input must be declared as an optional type, using the ? modifier. Outside of the if() block, stepB’s output is not guaranteed to exist, so stepC has to handle the possibility that it did not run by allowing that input to be optional.
Concrete example
Here we declare GVCFmode, a variable of the type Boolean. If it is true, then we want to run the tool in GVCF mode, otherwise we want to run it in normal mode. Essentially, this workflow allows you to select which HaplotypeCaller method you wish to run.
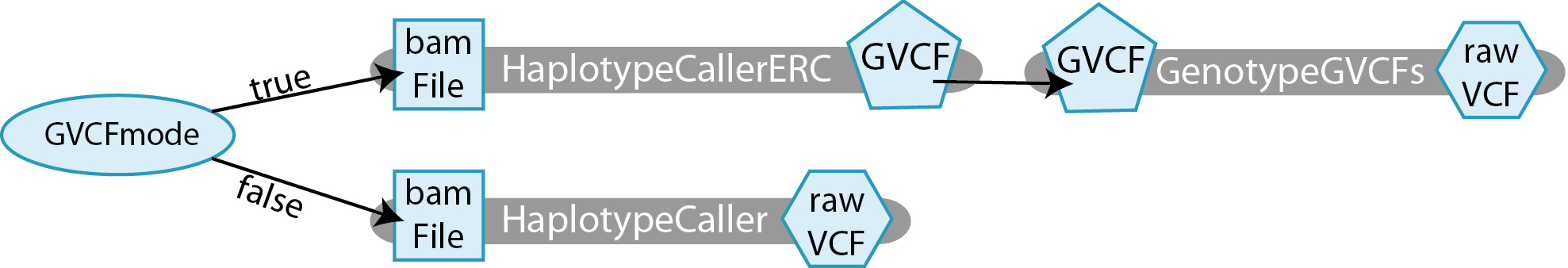
For our use case, we have declared three tasks, as follows:
- HaplotypeCallerERCtakes in a
File bamFileand produces aFile GVCF. - GenotypeGVCFs takes in a
File GVCFand produces aFile rawVCF. - HaplotypeCaller takes in a
File bamFileand produces aFile rawVCF.
Concrete example script
version 1.0
workflow ConditionalExample {
input {
Boolean GVCFmode
File inputBam
}
if (GVCFmode) {
call HaplotypeCallerERC {
input:
bamFile = inputBam
}
call GenotypeGVCF {
input:
GVCF = HaplotypeCallerERC.GVCF
}
}
if (!GVCFmode) {
call HaplotypeCaller {
input:
bamFile = inputBam
}
}
}
task HaplotypeCaller {
input {
File bamFile
}
command <<<
java -jar GenomeAnalysisTK.jar \
-T HaplotypeCaller \
-R reference.fasta \
-I ~{bamFile} \
-o rawVariants.vcf
>>>
output {
File rawVCF = "rawVariants.vcf"
}
}
task HaplotypeCallerERC {
input {
File bamFile
}
command <<<
java -jar GenomeAnalysisTK.jar \
-T HaplotypeCaller \
-ERC GVCF \
-R reference.fasta \
-I ~{bamFile} \
-o rawLikelihoods.gvcf
>>>
output {
File GVCF = "rawLikelihoods.gvcf"
}
}
task GenotypeGVCF {
input {
File GVCF
}
command <<<
java -jar GenomeAnalysisTK.jar \
-T GenotypeGVCFs \
-R reference.fasta \
-V ~{GVCF} \
-o rawVariants.vcf
>>>
output {
File rawVCF = "rawVariants.vcf"
}
}