Linear chaining
The simplest way to chain tasks together in a workflow is a linear chain, where we feed the output of one task to the input of the next, as shown in the diagram below.
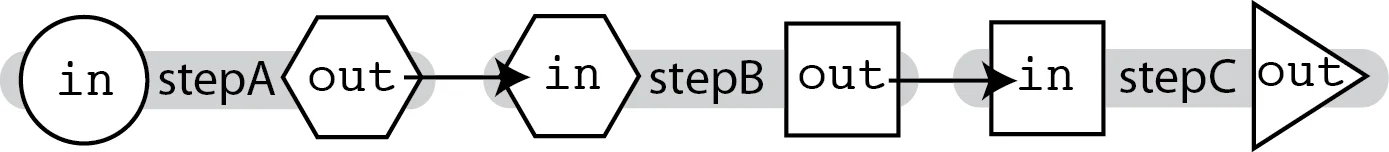
This is easy to do because WDL allows us to refer to the output of any task (declared appropriately in the task's output block) within the call statement of another task (and indeed, anywhere else in the workflow block), using the syntax task_name.output_variable.
Using the diagram as an example, we can specify in the call to stepB that we want it to use stepA.out as the value of the input variable, which we'll define as in. It's the same rule for stepC.
To write this in a WDL script, we'd use the following code:
call stepB {
input:
in = stepA.out
}
call stepC {
input:
in = stepB.out
}
This relies on a principle called "hierarchical naming" that allows us to identify components by their parentage.
Generic example script
To put this in context, here is what the code for the workflow illustrated above would look like in full:
version 1.0
workflow LinearChain {
input {
File firstInput
}
call stepA {
input:
in = firstInput
}
call stepB {
input:
in = stepA.out
}
call stepC {
input:
in = stepB.out
}
}
task stepA {
input {
File in
}
command <<<
programA I=~{in} O=outputA.ext
>>>
output {
File out = "outputA.ext"
}
}
task stepB {
input {
File in
}
command <<<
programB I=~{in} O=outputB.ext
>>>
output {
File out = "outputB.ext"
}
}
task stepC {
input {
File in
}
command <<<
programC I=~{in} O=outputC.ext
>>>
output {
File out = "outputC.ext"
}
}
Concrete example
Let’s look at a concrete example of linear chaining in a workflow designed to pre-process some DNA sequencing data (MarkDuplicates), perform an analysis on the pre-processed data (HaplotypeCaller), then subset the results of the analysis (SelectVariants).

The workflow involves three tasks:
- MarkDuplicates takes in a
File bamFileand produces aFile metricsand aFile dedupBAM. - HaplotypeCaller takes in a
File bamFileand produces aFile rawVCF. - SelectVariants takes in a
File VCFand aString typeto specify whether to select INDELs or SNPs. It produces aFile subsetVCF, containing only variants of the specified type.
Concrete example script
This is what the code for the workflow illustrated above would look like:
version 1.0
workflow LinearChainExample {
input {
File originalBAM
}
call MarkDuplicates {
input:
bamFile = originalBAM
}
call HaplotypeCaller {
input:
bamFile = MarkDuplicates.dedupBam
}
call SelectVariants {
input:
VCF = HaplotypeCaller.rawVCF,
type="INDEL"
}
}
task MarkDuplicates {
input {
File bamFile
}
command <<<
java -jar picard.jar MarkDuplicates \
I=~{bamFile} O=dedupped.bam M= dedupped.metrics
>>>
output {
File dedupBam = "dedupped.bam"
File metrics = "dedupped.metrics"
}
}
task HaplotypeCaller {
input {
File bamFile
}
command <<<
java -jar GenomeAnalysisTK.jar \
-T HaplotypeCaller \
-R reference.fasta \
-I ~{bamFile} \
-o rawVariants.vcf
>>>
output {
File rawVCF = "rawVariants.vcf"
}
}
task SelectVariants {
input {
File VCF
String type
}
command <<<
java -jar GenomeAnalysisTK.jar \
-T SelectVariants \
-R reference.fasta \
-V ~{VCF} \
--variantType ~{type} \
-o rawIndels.vcf
>>>
output {
File subsetVCF = "rawIndels.vcf"
}
}
Note that here for simplicity we omitted the handling of index files, which has to be done explicitly in WDL.